Cournia Lab Webservers & Workflows
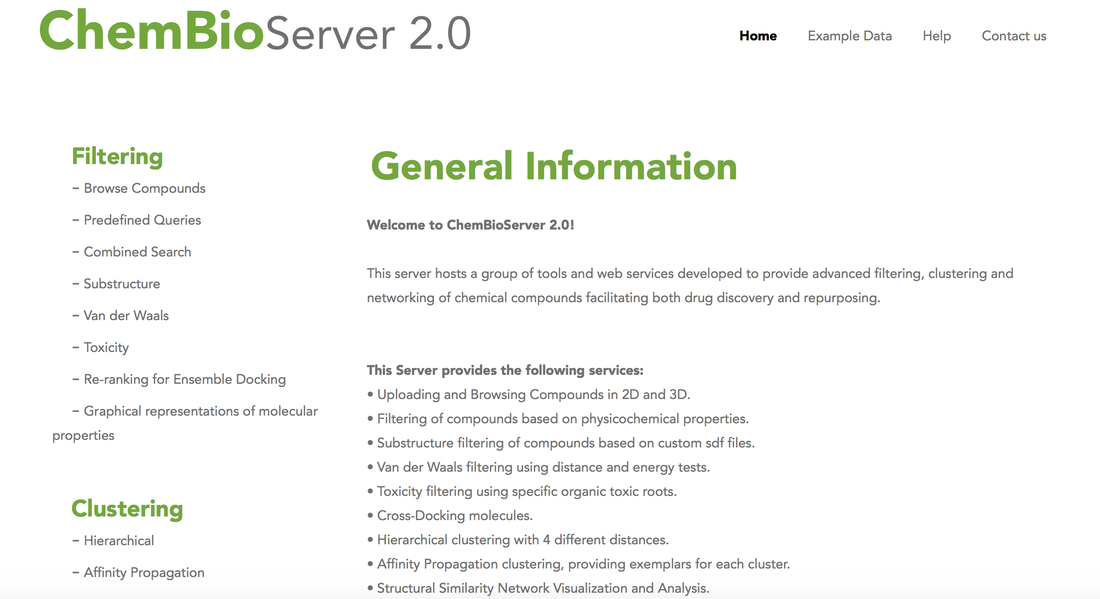
ChemBioServerChemBioServer is a publicly available web-application for effectively mining and filtering chemical compounds used in drug discovery. Features: (i) browse and visualize compounds along with their properties, (ii) filter chemical compounds for a variety of properties such as steric clashes and toxicity, (iii) apply perfect match substructure search, (iv) cluster compounds according to their physicochemical properties providing representative compounds for each cluster, (v) build custom compound mining pipelines and (vi) quantify through property graphs the top ranking compounds in drug discovery procedures. ChemBioServer allows for pre-processing of compounds prior to an in silico screen, as well as for post-processing of top-ranked molecules resulting from a docking exercise with the aim to increase the efficiency and the quality of compound selection that will pass to the experimental test phase.
|

NanoCrystal NanoCrystal is a novel web-based crystallographic tool that creates nanoparticle models from any crystal structure guided by their preferred equilibrium shape under standard conditions according to the Wulff morphology (crystal habit). Users can upload a cif file, define the Miller indices and their corresponding minimum surface energies according to the Wulff construction of a particular crystal, and specify the size of the nanocrystal. As a result, the nanoparticle is constructed and visualized, and the coordinates of the atoms are output to the user.
|
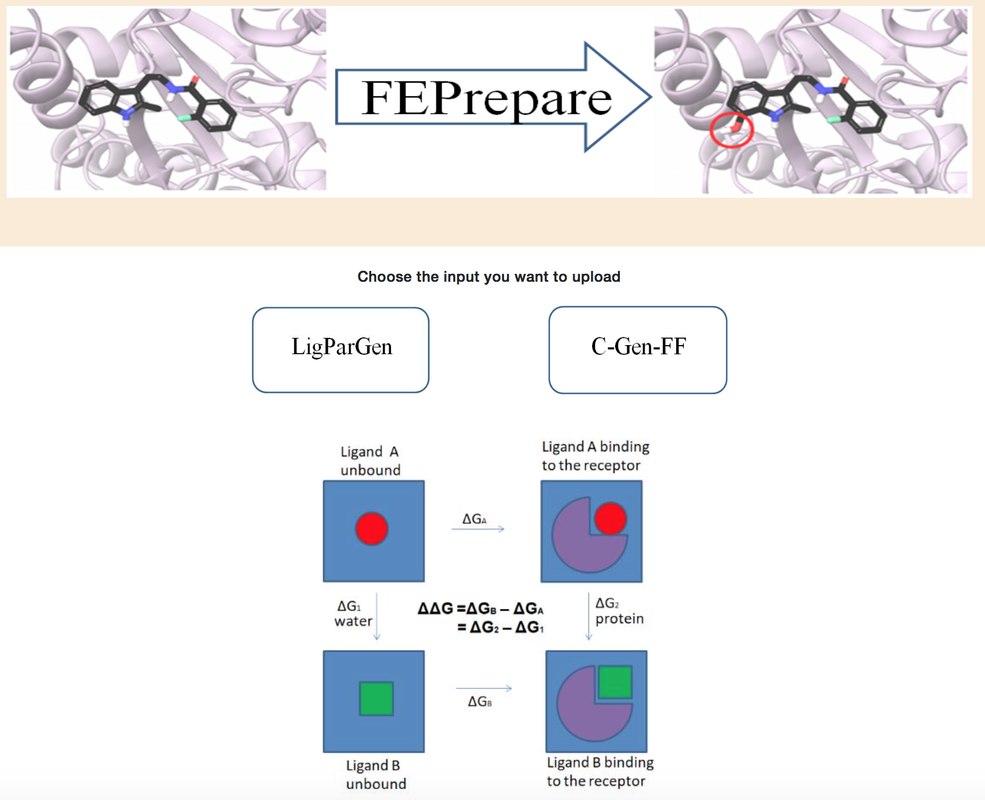
FEPrepareFEP prepare is a tool, which automates the set-up procedure for performing NAMD/FEP simulations. Automating free energy perturbation calculations is a step forward to delivering high throughput calculations for accurate predictions of relative binding affinities before a compound is synthesized, and consequently save enormous time and cost.
|
DREAMMDREAMM is a a web-based tool for the prediction of protein-membrane interfaces using ensemble machine learning. The allosteric modulation of peripheral membrane proteins (PMPs) by targeting protein-membrane interactions with drug-like molecules represents a new promising therapeutic strategy for proteins currently considered undruggable. However, the accessibility of protein-membrane interfaces by small molecules has been so far unexplored, possibly due to the complexity of the interface, the limited protein-membrane structural information and the lack of computational workflows to study it. Herein, we present a pipeline for drugging protein-membrane interfaces using the DREAMM (Drugging pRotein mEmbrAne Machine learning Method) web server. DREAMM works in the back end with a fast and robust ensemble machine learning algorithm for identifying protein-membrane interfaces of PMPs. Additionally, DREAMM also identifies binding pockets in the vicinity of the predicted membrane-penetrating amino acids in protein conformational ensembles provided by the user or generated within DREAMM.
|
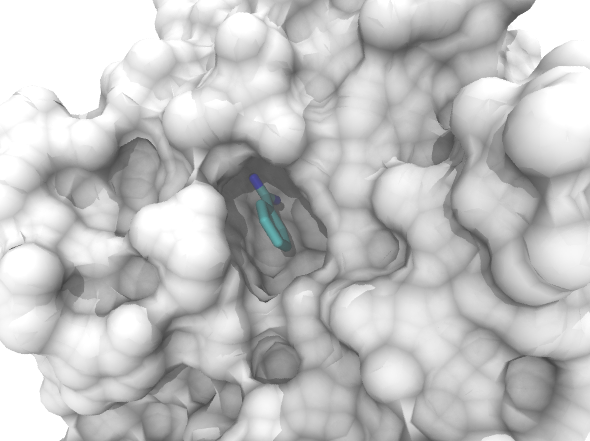
SubtractSubtract accurately calculates the volume of protein binding sites, and works both for crystal structures downloaded from the Protein Data Bank and for protein structures arising from Molecular Dynamics simulations trajectories.
Subtract accepts an atom selection in the form of a PDB file and computes the three- dimensional convex hull of the atoms points with the help of SciPy library. The next step of the algorithm is to compute the volume of the convex hull and the volume of the atoms that are included in the solid based on their van der Waals radii. The subtraction of those two volumes yields the volume of the investigated cavity. The algorithm computes cavity volumes of trajectory frames in parallel for maximum efficiency and speed. It requires minimal usage of memory due to the fact that it follows a buffering strategy of reading file chunks and therefore there is no need to load the entire file into memory. There is a wide support of trajectory formats like Gromacs trajectory files and multi-model PDB files due to its dependency to the MDTraj library. |
