Before starting to add elements to your PowerPoint file, the options of the file in question must be appropriately set to save all pasted images in their original resolution and high fidelity. If you have a PowerPoint file with images in it already, change the file options and then import/paste the original images again into the document to preserve their original resolution, as the images that were imported before setting these quality options have already been compressed.
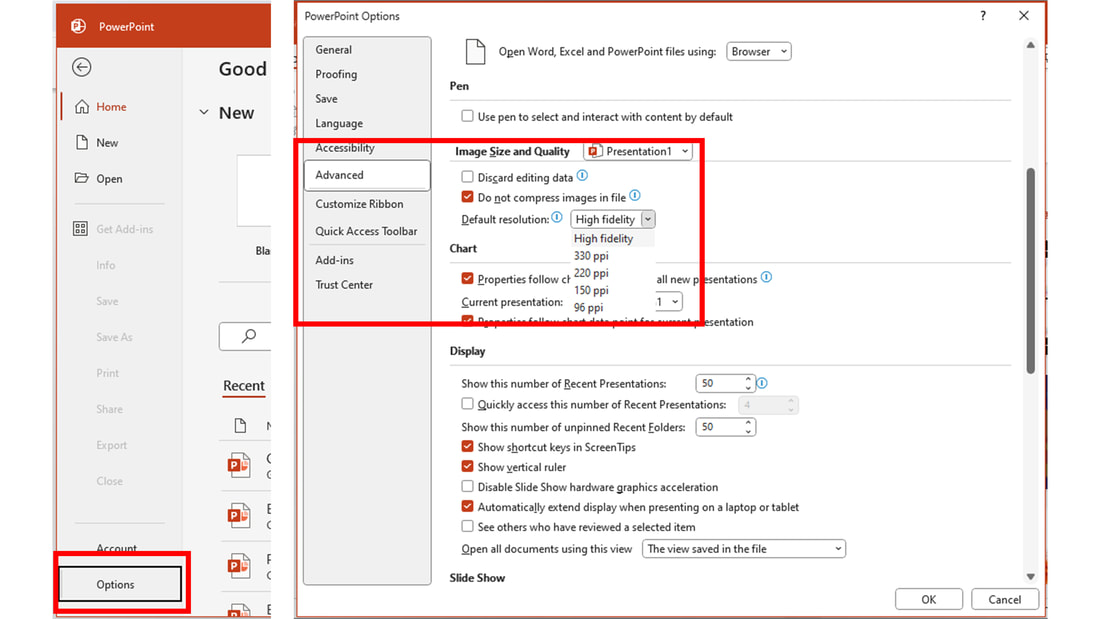
To change the file options for original image resolution and high fidelity, go to “Options” and make sure the following options are selected in the “Image Size and Quality” of the “Advanced” tab:
To change the file options for original image resolution and high fidelity, go to “Options” and make sure the following options are selected in the “Image Size and Quality” of the “Advanced” tab:
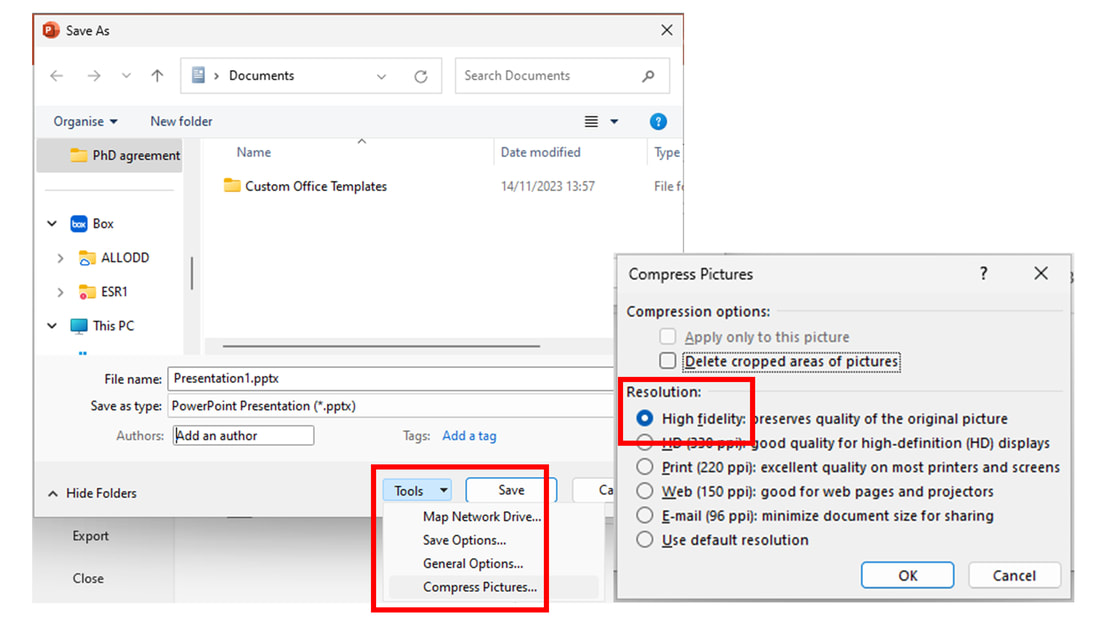
Finally, remember to double-check that you are saving/exporting your PowerPoint file to other formats maintaining the image quality. When “Saving as”, select “Tools” → “Compress pictures” options and ensure that “High fidelity” is selected before saving.
P.S.: to interconvert between image formats without losing quality, my first choice is https://www.zamzar.com/, although it has some size/amount of conversions limitations in its free version.
Francho Nerín Fonz
Francho Nerín Fonz
 RSS Feed
RSS Feed
